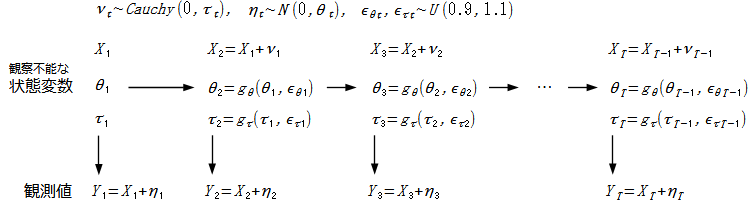BEEP音を鳴らしたかったのですが、検索するとbeeprパッケージを使え、alarm関数を使えと言うコレじゃないソリューションや、system関数でOSのコマンドを叩けと言うベタな手が引っかかります。どうやらRでBEEP音を鳴らしたい人々はほとんどいないようです。
Cの標準関数でBEEP音がサポートされていないせいか、Rは標準ではBEEP音を鳴らせません。その他の音も鳴らせません。パッケージの力を借りる事になるのですが、audioパッケージのplay関数が正弦波を音に変えてくれるので、BEEP音の代わりに使えます。
早速、どれみふぁそらしど…とベタに鳴らしてみましょう。
library(audio)
mkHz <- function(h = 4){
Hz <- 440*(2^(1/12))^{(-12*4):(12*h)}
n <- length(Hz)
scale <- c("C", "C#", "D", "D#", "E", "F", "F#", "G", "G#", "A", "A#", "B")
j <- 1
lst <- list()
while(0 < n){
lst[[j]] <- paste(scale[1:min(n, 12)], j, sep="")
j <- j + 1
n <- n - 12
}
names(Hz) <- unlist(lst)
Hz
}
mkWave <- function(..., length, volume, rate){
if(0 >= ...length()) stop("no scale inputed!")
Hz <- mkHz()
lst <- list()
for(i in 1:length(...elt(1))){
sinc <- 0
for(j in 1:...length()){
s <- ...elt(j)[i]
if(!is.na(Hz[s])){
sinc <- sinc + sin(1:rate/rate*(2*pi)*Hz[s])
}
}
sinc <- sinc / ...length()
lst[[i]] <- (sinc * volume[i])[1:(length[i])]
}
unlist(lst)
}
rate <- 44100
scale <- c("C5", "D5", "E5", "F5", "G5", "A5", "B5", "C6")
length <- rep(rate/4, length(scale))
volume <- rep(1/4, length(scale))
wait(play(mkWave(scale, length = length, volume = volume, rate = rate), rate))
正弦波を加算すれば和音(コード)になりますし、原理的には色々とできるわけですが、MIDIの再発明みたいな事になるのでこの辺でやめておこうかと思っていたのですが、正解/不正解/挨拶の効果音を用意しました。
correct <- function(rate = 44100){
scale <- c("E5", "C5", "E5", "C5", "E5", "C5")
length <- c(rep(rate/8, length(scale)-1), rate/4)
volume <- rep(1/4, length(scale))
wait(play(mkWave(scale, length = length, volume = volume, rate = rate), rate))
}
wrong <- function(rate = 44100){
scale1 <- c("F#3", "F#3")
scale2 <- c("G2", "G2")
length <- c(rate/3, rate/1.5)
volume <- rep(2/3, length(scale1))
wait(play(mkWave(scale1, scale2, length = length, volume = volume, rate = rate), rate))
}
bow <- function(rate = 44100){
length <- c(rate, rate, rate)
volume <- rep(2/3, 3)
wait(play(mkWave(
c("C3", "G2", "C2"),
c("C4", "G3", "C3"),
c("E4", "D4", "E4"),
c("G4", "F4", "G4"),
c("C5", "G4", "C5"),
c("", "B3", ""),
length = length, volume = volume, rate = rate), rate))
}
fanfare <- function(rate = 44100){
scale <- c("F4", "A4", "D5", "F5", "", "D5", "F5")
length <- c(rep(1/5, 6), 1/2)*rate
volume <- rep(1/2, length(scale))
wait(play(mkWave(scale, length = length, volume = volume, rate = rate), rate))
}
bigben <- function(rate = 44100){
scale <- c("F4", "A4", "G4", "C4", "F4", "G4", "A4", "F4", "A4", "F4", "G4", "C4", "C4", "G4", "A4", "F4")
length <- rep(c(1, 1, 1, 2), 4) * rate
volume <- rep(1/2, length(scale))
wait(play(mkWave(scale, length = length, volume = volume, rate = rate), rate))
}
bow()
fanfare()
correct()
wrong()
bigben()